
14/12/2021 - Nota de prensa
Este nuevo recurso, desarrollo por el equipo del Grupo de descubrimiento de fármacos basados en GPCRs del Instituto Hospital del Mar de Investigaciones Médicas, permite analizar en tres dimensiones los movimientos de las proteínas que forman parte del virus que provoca la COVID-19. Esto puede ayudar a los investigadores a entender su funcionamiento y a desarrollar nuevos tratamientos y vacunas
La herramienta, disponible en línea para todos los investigadores, proporciona un gran número de simulaciones del funcionamiento de estas proteínas, así como recursos para predecir cómo puede cambiar su función en relación con las mutaciones que se pueden producir en la estructura de este coronavirus
Los impulsores de la iniciativa han utilizado más de 360 gigabits de datos para llevarla a cabo. Es la única base de datos creada hasta ahora que combina simulaciones de proteínas con datos de mutaciones para estudiar al SARS-CoV-2
Los investigadores disponen de una nueva herramienta para hacer frente al SARS-CoV-2, analizarlo y buscar nuevas formas para luchar contra la COVID-19. Se trata de la base de datos SCoV2-MD (que se puede consultar en www.scov2-md.org) que contiene información detallada, a nivel atómico, dinámico y en tres dimensiones, de todas las proteínas con estructura tridimensional conocida de este coronavirus. En total, contiene 360 gigabits de datos sobre la mayoría de las 29 proteínas que forman parte de él: cuatro estructurales, 16 no-estructurales y nueve accesorias. Nucleic Acids Research ha publicado un artículo sobre su funcionamiento, que ha considerado uno de los más destacados entre los artículos publicados en la revista.
Las proteínas son moléculas básicas en el funcionamiento de las células. En el caso del SARS-CoV-2, el coronavirus responsable de la COVID-19, son las responsables de su capacidad de infectar a los seres humanos, así como de su propagación. Es el caso de la llamada proteína espiga, que forma la característica corona que da su nombre este tipo de virus. La nueva base de datos, impulsada por el Grupo de descubrimiento de fármacos basados en GPCRs del Instituto Hospital del Mar de Investigaciones Médicas (IMIM-Hospital del Mar), en colaboración con el Biophysics Institute (CNR-IBF) del National Research Council de Italia, el Paul Scherrer Institute de Suiza y Dompé Farmaceutici de Italia, permite conocer con un grado de detalle nunca visto su estructura y su funcionamiento, así como predecir su evolución a lo largo de las diferentes mutaciones que ha sufrido y que sufrirá el virus.

De izquierda a derecha: Jana Selent y Mariona Torrens-Fontanals.
Herramienta a disposición de todos los investigadores
Esta herramienta, una de las más potentes creadas hasta ahora combinando simulaciones de la estructura tridimensional de las proteínas con datos de mutaciones en el virus responsable de la COVID-19, está disponible para cualquier investigador, en línea y de forma gratuita. Al conectarse, sin necesidad de utilizar ninguna aplicación específica, pueden ver la estructura de las proteínas del SARS-CoV-2 a nivel atómico, gracias al análisis realizado por los creadores de la base de datos a partir de la información generada por ellos mismos y la disponible en diferentes repositorios públicos. Para hacerlo, se han utilizado herramientas computacionales de simulación de la dinámica molecular, que permiten hacer predicciones de cómo cada átomo de la proteína actuará siguiendo las leyes de la física, para construir así un modelo tridimensional del comportamiento de esta molécula. Las simulaciones acumuladas, 252 en total, también prevén el impacto de las mutaciones conocidas del coronavirus sobre las proteínas.
"Lo que realmente es nuevo es que utilizamos datos dinámicos combinados con la evolución del virus para predecir su impacto en la función de las proteínas", explica Jana Selent, investigadora del IMIM-Hospital del Mar y autora principal del artículo sobre esta nueva herramienta. Para hacerlo, "más allá de las simulaciones del comportamiento de las proteínas, se ha tenido en cuenta la información disponible sobre la genética del virus para calcular y predecir el impacto de las mutaciones", añade Toni Giorgino, coautor principal del artículo. Por este motivo, SCoV2-MD se convierte en una herramienta útil para visualizar cómo estas mutaciones del SARS-CoV-2 afectarán a su capacidad de transmisión y para infectar las células humanas a través de los cambios que producen en las proteínas que formen parte de él.
A la vez, esta nueva herramienta también puede ayudar a los investigadores a desarrollar nuevos tratamientos y vacunas contra la COVID-19. "Ver las simulaciones permite ver y entender cómo se comporta, cómo funciona y qué partes de la estructura de la proteína son importantes y posibles dianas para el estudio de nuevos tratamientos", apunta Mariona Torrens-Fontanals, también investigadora del IMIM-Hospital del Mar y primera firmante del trabajo. Incluso, facilita la posibilidad de predecir si las mutaciones del coronavirus pueden afectar la capacidad de los anticuerpos que forman las vacunas contra la COVID-19 para reconocer el virus y activar el sistema inmunitario contra él.
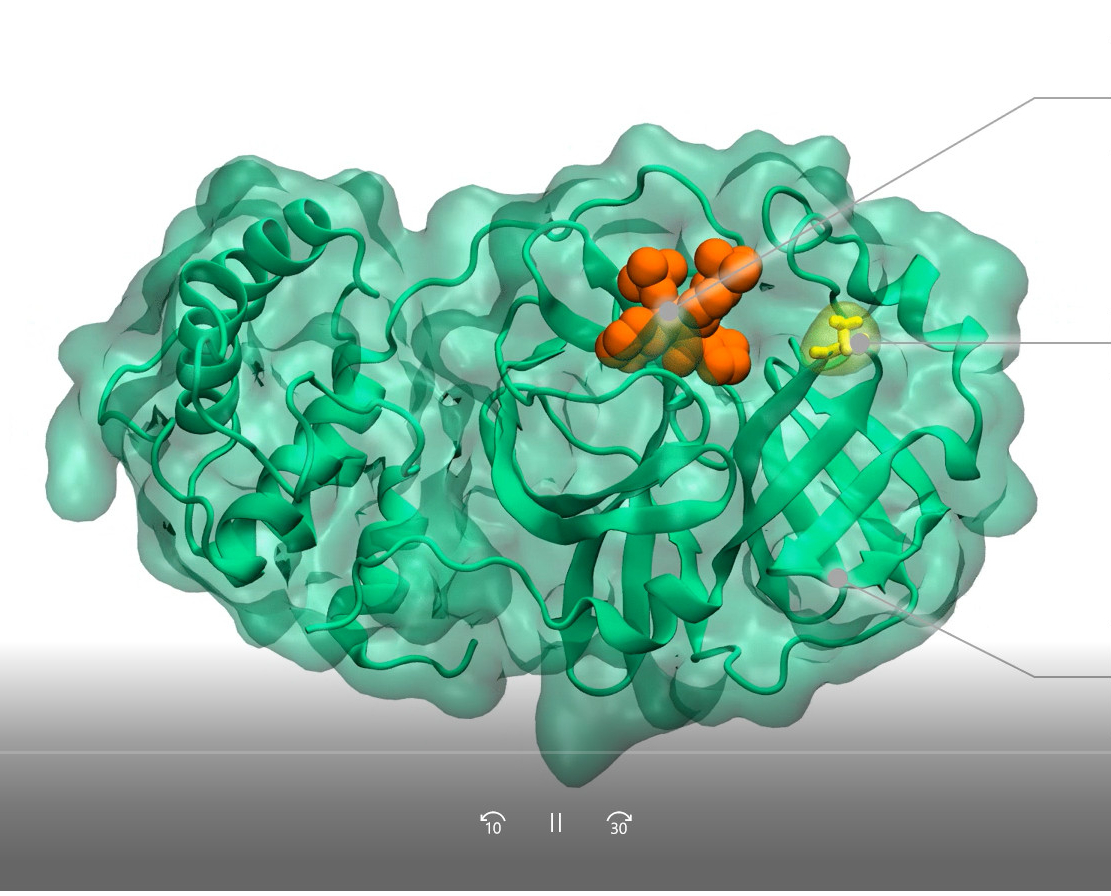
El vídeo muestra que al combinar información de la dinámica estructural de proteínas y datos de la evolución del virus, SCoV2-MD permite estudiar el impacto de mutaciones específicas (por ejemplo, T24A en la proteasa principal del SARS-CoV-2) en la unión de compuestos antivirales (por ejemplo, el inhibidor ML188).
Artículo de referencia
Mariona Torrens-Fontanals, Alejandro Peralta-García, Carmine Talarico, Ramon Guixà-González, Toni Giorgino, Jana Selent, SCoV2-MD: a database for the dynamics of the SARS-CoV-2 proteome and variant impact predictions, Nucleic Acids Research, 2021; https://doi.org/10.1093/nar/gkab977
Servei de Comunicació:
Marta Calsina(ELIMINAR)
Tel:
(+34) 93 316 06 80
Doctor Aiguader, 88
08226 Barcelona
© Institut Hospital del Mar
d'Investigacions MèdiquesAviso legal y Política de Privacidad | Política de cookies | Mapa Web | Accesibilidad | Dirección y accesos | Contacto
